
Featured Publications
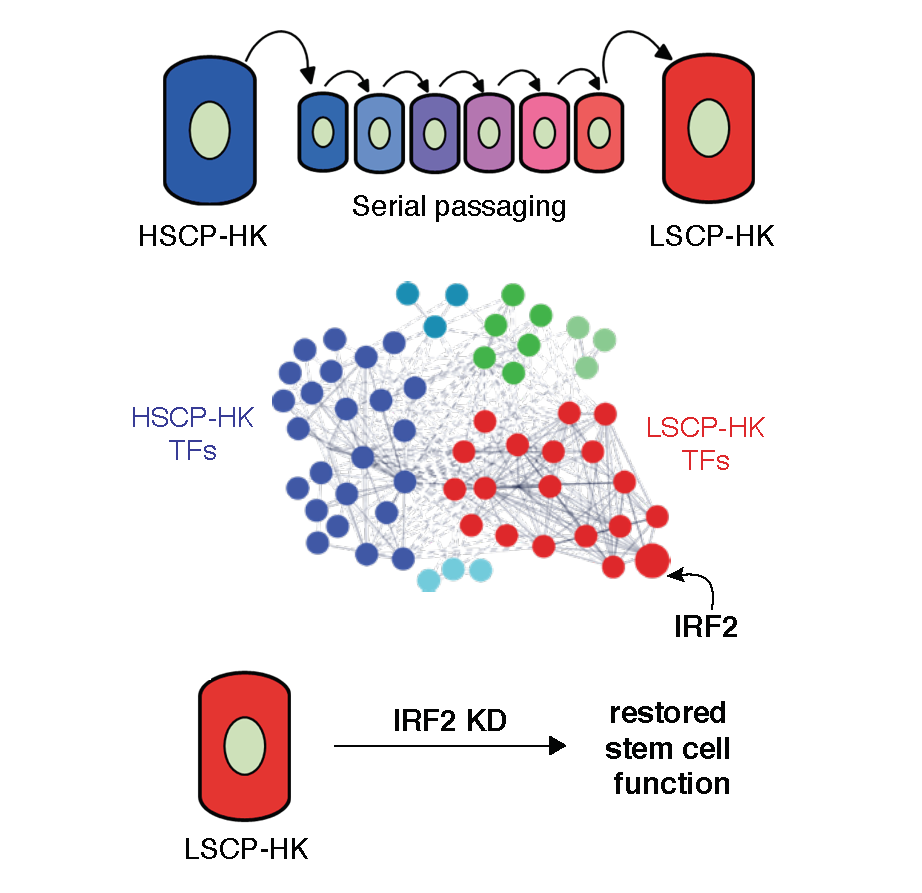
IRF2 IS A MASTER REGULATOR OF HUMAN KERATINOCYTE STEM CELL FATE (MERCADO ET AL., 2019 NATURE COMMUNICATIONS)
Resident adult epithelial stem cells maintain tissue homeostasis by balancing self-renewal and differentiation. The stem cell potential of human epidermal keratinocytes is retained in vitro but lost over time suggesting extrinsic and intrinsic regulation. Transcription factor-controlled regulatory circuitries govern cell identity, are sufficient to induce pluripotency and transdifferentiate cells. We investigate whether transcriptional circuitry also governs phenotypic changes within a given cell type by comparing human primary keratinocytes with intrinsically high versus low stem cell potential. Using integrated chromatin and transcriptional profiling, we implicate IRF2 as antagonistic to stemness and show that it binds and regulates active cis-regulatory elements at interferon response and antigen presentation genes. CRISPR-KD of IRF2 in keratinocytes with low stem cell potential increases self-renewal, migration and epidermis formation. These data demonstrate that transcription factor regulatory circuitries, in addition to maintaining cell identity, control plasticity within cell types and offer potential for therapeutic modulation of cell function.

Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma (mack et al., 2019 Journal of Experimental Medicine)
Glioblastoma (GBM) remains today an incurable and highly aggressive brain tumor. Large consortia based efforts have sought to define, dissect, and catalogue the landscape of mutations of GBM, with the goal of pairing mutated cancer genes with therapeutic targets. Unlike other cancers, these approaches have failed to reveal actionable targets for therapy in GBM; thus calling for alternative strategies to discover new drugs. Here, we mapped active chromatin landscapes with gene expression, whole-exomes, copy number profiles, and DNA methylomes across 44 glioblastoma stem cell (GSCs) models, 50 primary tumors, and 10 neural stem cells (NSCs) to identify essential super enhancer (SE)-associated genes and the core transcription factors (TFs) that establish SEs and maintain GSC identity. We show that GSCs segregate into two groups dominated by distinct enhancer profiles and unique developmental core TF regulatory programs. Group specific TFs enforce GSC identity — they exhibit higher activity in GBM versus normal NSCs, are associated with poor clinical outcomes, and are required for GBM growth in vivo. Although TFs are commonly considered undruggable, we show that group specific enhancer regulation of the MAPK/ERK pathway predicts sensitivity to MEK inhibition. These data demonstrate that transcriptional identity can be leveraged to identify novel dependencies and therapeutic approaches.

Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype (olson et al., 2019 cell chemical biology)
Cyclin-dependent kinase 7 (CDK7) is a key regulator of both the cell cycle and transcription, but its precise role remains uncertain. THZ1, a CDK7 inhibitor, was previously shown to strongly inhibit superenhancer-associated gene expression. However, potent CDK12/13 off-target activity obscured CDK7’s contribution to the initially observed phenotype. Here, we characterize a newly discovered, more selective covalent inhibitor of CDK7 in YKL-5-124. Treatment with this compound induces G1 arrest and inhibits E2F-driven cell cycle genes, phenotypes which are furthermore rescued by a CDK7 mutant which renders resistance to engagement by YKL-5-124. Unlike THZ1, treatment with YKL-5-124 resulted in no change to RNA polymerase II CTD phosphorylation. Concurrent treatment with YKL-5-124 and THZ531, a selective CDK12/13 inhibitor, was able to phenocopy THZ1. These findings highlight the importance of CDK7/12/13 polypharmacology for the anti-cancer activity of THZ1 and posits that CDK7 inhibition may be useful in the treatment of cancers characterized by E2F misregulation.

Small-molecule targeting of brachyury transcription factor addiction in chordoma (Sharifnia et al., 2019 Nature medicine)
Chordoma is a primary bone cancer with no approved therapy. Here we describe the discovery of therapeutically targetable chordoma dependencies via genome-scale CRISPR-Cas9 screening and focused small-molecule sensitivity profiling. These systematic approaches reveal that the developmental transcription factor T (brachyury; TBXT) is the top selectively essential gene in chordoma, and that transcriptional cyclin dependent kinase (CDK) inhibitors targeting CDK7/12/13 and CDK9 potently suppress chordoma cell proliferation. In chordoma, we find that T is associated with a 1.5-Mb region containing a cluster of super enhancers and is the most highly expressed super-enhancer associated transcription factor. Transcriptional CDK inhibition leads to preferential downregulation of brachyury protein levels in all models tested, and, in vivo, CDK7/12/13-inhibitor treatment substantially reduces tumor growth. Together, these data demonstrate small-molecule targeting of brachyury transcription factor addiction in chordoma, identify a mechanism of T gene regulation that underlies this therapeutic strategy, and provide a blueprint for applying systematic genetic and chemical screening approaches to discover vulnerabilities in genomically quiet cancers.

Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma (Zeid et al., 2018 Nature Genetics)
Neuroblastoma is one of the most frequent pediatric solid tumors. Amplification of the transcription factor MYCN is the central hallmark of high-risk disease and results in excess levels of the protein in the nucleus where it can bind many thousands of regions the genome. Here we show that when amplified, oncogenic MYCN begins to invade tissue specific enhancers where it reshapes the neuroblastoma gene expression program in collaboration with tissue specific transcription factors like TWIST1. This axis of enhancer regulated genes correlates with poor survival and is dependent on tissue specific transcription factor activity creating an avenue to selectively target the activity of oncogenic MYCN.
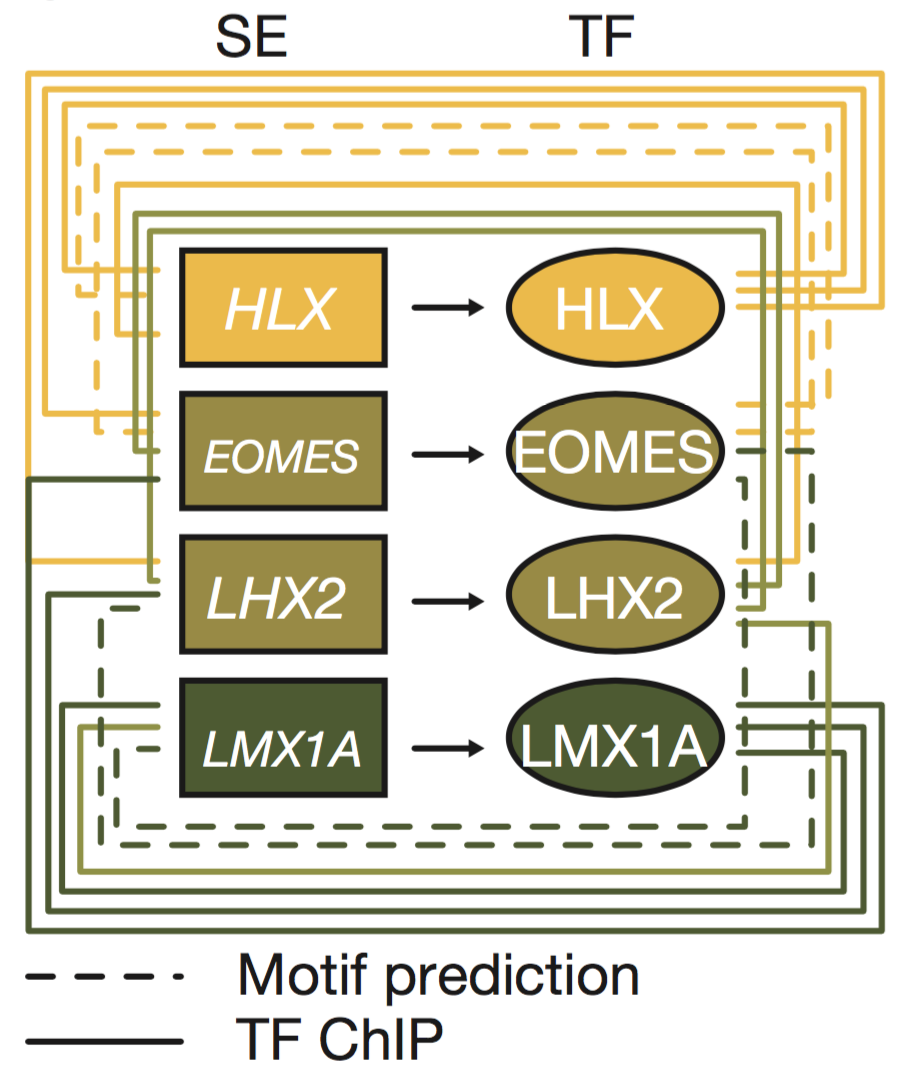
Medulloblastoma origins (Lin et al., 2016 Nature)
In this collaboration with St. Jude's and the German cancer research center (DKFZ), we mapped active enhancer landscapes across all four known medulloblastoma subtypes. In many developmental models including embryonic stem cells, large clustered enhancer elements (super-enhancers or SEs) regulate key lineage specifying transcription factors that in turn bind and regulate other lineage transcription factors through enhancers. We hypothesized and showed that an inference of transcription factor connectivity learned through analyzing subtype specific enhancer landscapes could reveal key subtype specific lineage master regulators. We identified a subset of transcription factors (HLX, EOMES, LHX2, and LMX1A) that delineate the Group3/Group4 regulatory axis. Lineage tracing of these factors identified spatiotemporal restricted expression in the nuclear transitory zone, an assembly point for immature deep cerebellar nuclei. Deletion of LMX1A, a Group4 specific factor, in mice causes developmental defects in the developing cerebellum in this region and reductions in expression for other Group4 genes. This approach establishes a framework for the inference of tumor cell of origin mapping through the analysis of enhancer regulator landscapes.